创新背景
计算蛋白质设计(CPD)是指通过计算理性地确定蛋白质的氨基酸序列,实现预设的结构和功能。计算蛋白质设计已经有了一套系统方法,得到诸多实验验证。这些方法既可用于从头设计蛋白,也可以用于既有蛋白的理性改造,具有广泛应用前景,是合成生物学的重要使能技术之一。现在,计算机蛋白质设计在医疗、生物工程等众多与人类健康息息相关的领域发挥作用。
创新过程
计算蛋白质设计依赖结构功能和蛋白序列的对应关系。蛋白质折叠是蛋白链以迅速和可再现的方式获得其天然的三维结构,三维结构由氨基酸序列或一级结构确定。逆向蛋白质折叠是现在CPD的主要研究方向之一,又名固定骨架设计,是反向生成给定的蛋白质结构的新氨基酸序列。
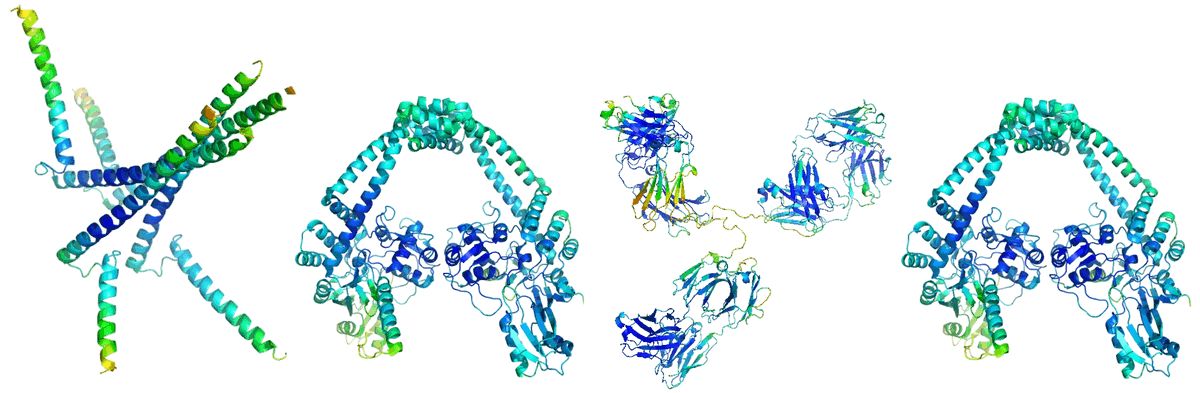
2022年4月15日,bioRxiv 预印平台发布了芝加哥丰田计算技术研究所的许锦波教授团队创建的一个你蛋白质折叠深度学习框架成果《一种用于学习逆蛋白质折叠的深度 SE(3)-等变模型》。模型能从结构数据中学习蛋白质功能,帮助进一步利用结构信息预测蛋白质的突变。
研究团队认为,如果能仅从结构数据中获得蛋白质功能有关的信息,那么仅以蛋白结构和部分序列为条件的生成模型,也可以用作一个对单点突变的功能影响的零样本预测器。

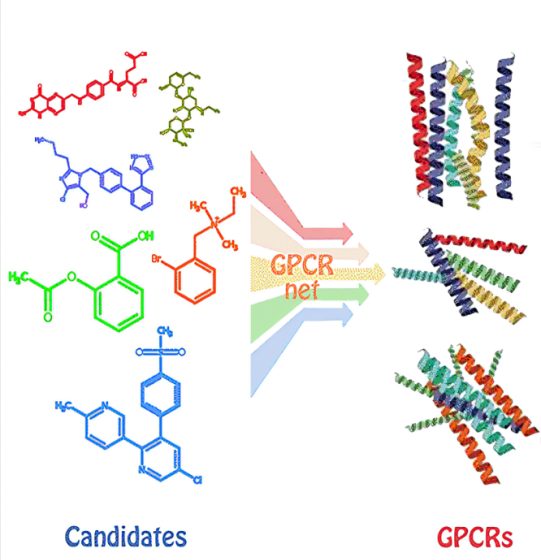
研究使用12 层 Locality Aware Graph Transformer 和 8 层 TFN-Transformer两个主要子模块组成的深度 SE(3)-等变图 transformer 架构,对从蛋白质主干结构派生的特征进行操作。然后比较现有的几种逆向折叠方法,证明新方法明显具有更高的天然序列恢复率(NSR)。将预测的突变概率与突变扫描结果比较,验证了新模型预测蛋白质的效果。结果表明,新方法的NSR比现有的同类模型要优异许多。
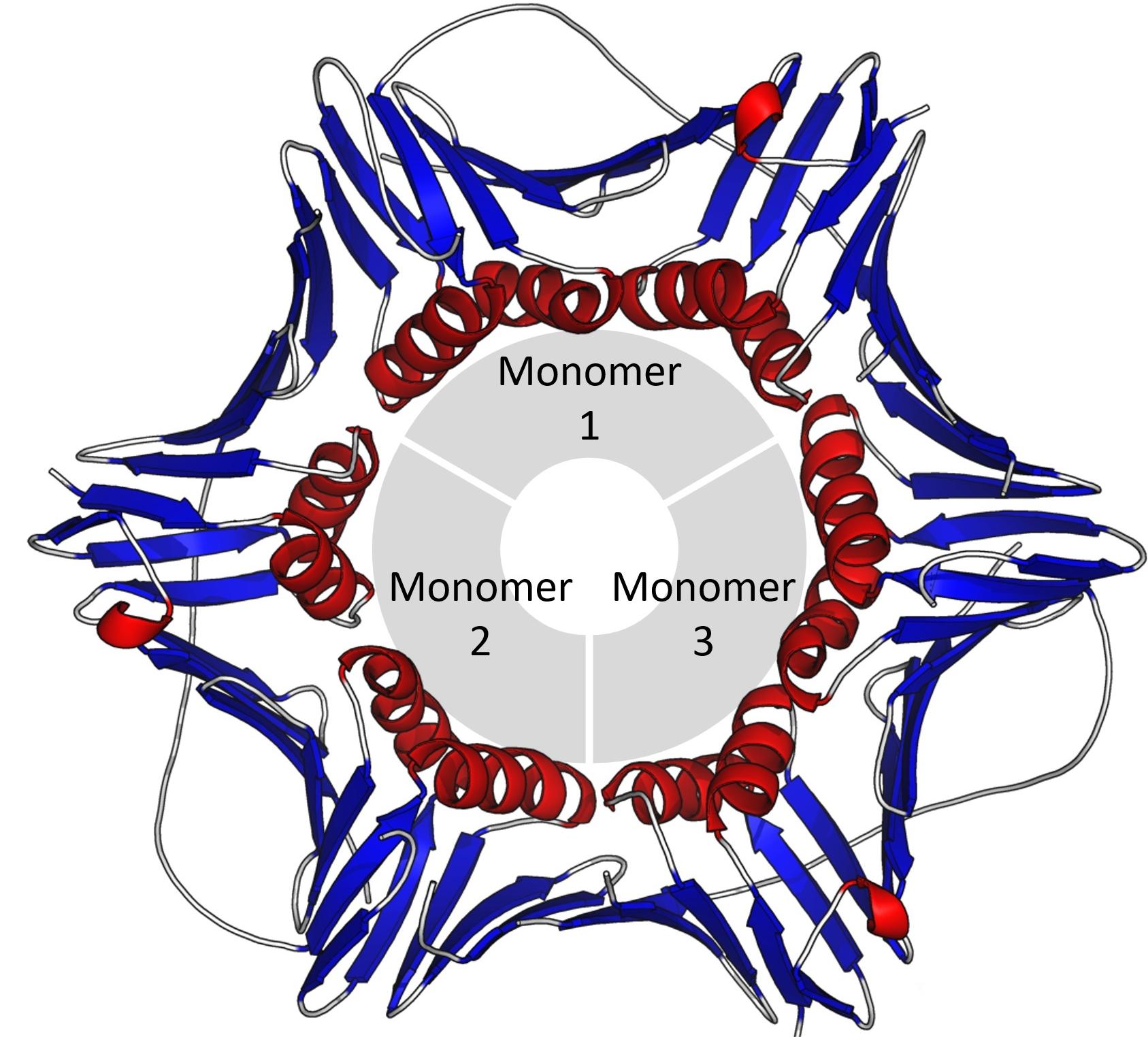
研究人员使用几个深度突变扫描(DMS)数据集的稳定性数据来比较预测出来的点突变的对数,以便更好地理解该模型设计捕获潜在蛋白质的功能。对比结果表明,新模型在DMS 数据集上测试蛋白质突变效应零样本预测器,优于大型序列数据库训练出来的蛋白质语言模型。说明新方法捕获了三维构象和氨基酸序列与功能的相互关系,并证明结构信息有助于表征蛋白质突变效应。
新模型结合你想蛋白质折叠的几何性质和等变神经网络及新颖的注意力机制,改进了现有的方法,使预测蛋白质序列的结果更加准确。并且新模型对蛋白质突变效应的预测更加准确,可以更快捕获蛋白质性能。研究将对和人类健康相关的生物科学、医学和酶工程等起到极大的促进作用。
创新关键点
创新结合逆向蛋白质折叠结构进性质和新的结构信息机制,开发新的深度学习模型。
The new reverse computational protein design framework improves sequence prediction speed and accuracy
Computational protein design relies on structure-function and protein-sequence correspondences. Protein folding is the rapid and reproducible acquisition of protein chains in their native three-dimensional structure, which is determined by amino acid sequence or primary structure. Reverse protein folding is one of the main research directions of CPD, also known as fixed backbone design, which is to generate a new amino acid sequence of a given protein structure in reverse.
On April 15, 2022, the bioRxiv preprint platform released the results of a deep learning framework for protein folding created by the team of Professor Jinbo Xu of the Toyota Institute of Computing Technology in Chicago, "A Deep SE(3)-Equivariant Model for Learning Inverse Protein Folding." Models can learn protein function from structural data and help further predict protein mutations using structural information.
The research team believes that generative models conditioned only on protein structure and partial sequence can also be used as a zero-sample predictor of the functional impact of single point mutations if information about protein function can be obtained only from structural data.
The study uses a deep SE(3)-equivariant graph transformer architecture composed of two main sub-modules, a 12-layer Locality Aware Graph Transformer and an 8-layer TFN-Transformer, to operate on features derived from the protein backbone structure. Several existing reverse-folding methods were then compared, demonstrating that the new method has a significantly higher native sequence recovery rate (NSR). Comparing the predicted mutation probabilities with the mutation scan results validated the effect of the new model for predicting proteins. The results show that the NSR of the new method is much better than the existing similar models.
The researchers used stability data from several Deep Mutation Scan (DMS) datasets to compare the logarithm of predicted point mutations to better understand how the model was designed to capture the function of the underlying protein. The comparison results show that the new model is better than the protein language model trained on the large sequence database in testing the zero-sample predictor of protein mutation effect on the DMS dataset. The new method captures the three-dimensional conformation and the relationship between amino acid sequence and function, and demonstrates that structural information is useful for characterizing protein mutation effects.
The new model combines the geometric properties of protein folding with equivariant neural networks and novel attention mechanisms, improving existing methods and making the results of predicting protein sequences more accurate. And the new model is more accurate in predicting the effect of protein mutations, and can capture protein performance faster. The research will greatly promote the biological sciences, medicine and enzyme engineering related to human health.
智能推荐
生命科学理论创新 | 果蝇内脏发现新型细胞死亡类型
2022-06-29从细胞死亡方式入手研究新型细胞死亡类型。
涉及学科涉及领域研究方向高龄父亲精子中miRNA影响胚胎,使子代易出现精神疾病
2022-08-03这项研究发现,高龄雄鼠精子中的miRNA通过引发早期胚胎雌二醇信号通路功能紊乱,导致子代出现焦虑和社交障碍等精神症状表型。该发现对阐释雄性高龄生育导致子代精神异常的分子机理具有重要意义,也为研究精神类疾病的遗传规律提供了新的视角;同时,在全球生育年龄普遍推迟的社会现实下,此发现对于预防子代可能出现的健康风险、维持优生优育的政策提供了一定的理论依据。
涉及学科涉及领域研究方向研究发现逆向TCA循环可将二氧化碳转化为氨基酸、糖和脂质
2022-08-04在细菌内发现逆转的三羧酸循环,逆向TCA循环令二氧化碳成为微生物碳源。
涉及学科涉及领域研究方向大脑前额叶皮层中有毒副产物的积累导致认知疲劳
2022-08-17巴黎 Pitié-Salpêtrière 医院和巴黎大脑研究所的研究团队经过两组对比实验分析发现,持续的高强度工作不仅会导致瞳孔扩张的减少,还会造成更高水平的谷氨酸积累。这项研究的结果否定了以往认为疲劳是大脑制造的幻觉的理论,表明了认知工作会导致真正的大脑功能的改变,即有害物质的积累。因此,疲劳确实会是一种让人停止工作的信号,但目的不同:它是为了保持大脑功能的完整性。
涉及学科涉及领域研究方向